Two-photon absorption (2PA) is an electronic excitation process involving the simultaneous absorption of two photons. 2PA probability is quadratically dependent on the intensity of incident light, so it may be localized in space with a tightly focused laser beam. There are a wide range of 2PA applications, such as lithographic micro-fabrication, three dimensional data storage, photonic devices, quantum information technology, optical limiting, two-photon pumped lasing in organic chromophores and quantum dots, in-vivo imaging, and cell-selective photo-dynamic therapy. Most applications require chromophores with large 2PA cross-sections to minimize laser intensity requirements and prevent overheating of targets. To design more efficient 2PA chromophores, it is important to understand their structure/activity relationships. Computer modeling of 2PA spectra facilitates understanding of these relationships and represents a rational approach in chromophore design.
Time-Dependent Density Functional Theory (TD-DFT) was recently combined with the Coupled Electronic Oscillator (CEO) formalism to simulate 2PA electronic spectra in large conjugated molecules [1, 2]. These and other 2PA predictions using TD-DFT [3] were shown to achieve superior accuracy, when compared to semiempirical wavefunction theory methods. In this contribution we calculate state-to-state transition dipole moments µinm using a posteriory Tamm-Dancoff approximation (ATDA, introduced in Ref. [4]) and employ them to identify the essential states governing the 2PA process. We also validate ATDA by using these approximate µinm to predict the resonant 2PA cross-sections with sum over state models (SOS) and compare them to CEO results as well as experimental values.
In the SOS/2PA transition matrix approximation the 2PA cross-section is given by [5]:
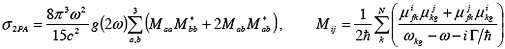
here g(2ω) is a Lorentzian lineshape and µinm are state-to-state transition dipole moments. ATDA/SOS approach opens new venues for interpretation of 2PA properties in terms of molecular electronic structure and can be used for rational design of 2PA chromophores. We present 2PA profiles obtained with both SOS and CEO formalisms on Fig.1. To analyze the electronic structure of the excited states, we use the natural transition orbitals, which diagonalize the transition density matrix and give the best representation of the electronic excitation in single-particle terms [6].
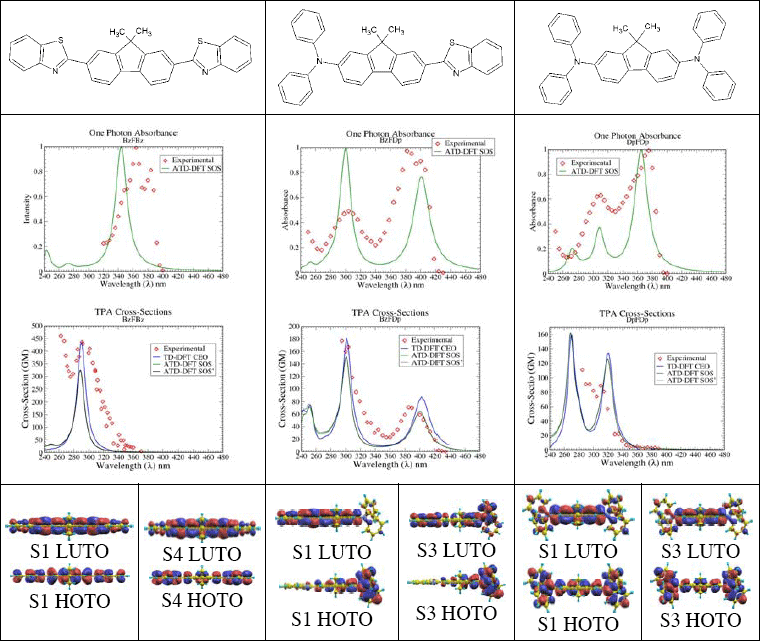
SOS and CEO results are in quantitative agreement with each other, which provides a validation for ATDA/SOS method. Predicted 2PA profiles also agree well with experimental ones. This validates the use of TD-DFT as a part of rational design strategies directed toward new and improved Two-Photon absorbing materials.
References
[1] A. E. Masunov, S. Tretiak, Journal of Physical Chemistry B 2004, 108, 899.
[2] E. A. Badaeva, T. V. Timofeeva, A. E. Masunov, S. Tretiak, Journal of Physical Chemistry A 2005, 109, 7276.
[3] P. Cronstrand, B. Jansik, D. Jonsson, Y. Luo, H. Agren, Journal of Chemical Physics 2004, 121, 9239.
[4] I. Mikhailov, S. Tafur, A. E. Masunov, Phys. Rev. A 2008, 77, 012510.
[5] K. Ohta, K. Kamada, Journal of Chemical Physics 2006, 124.
[6] R. L. Martin, Journal of Chemical Physics 2003, 118, 4775.