The donor and/or acceptor (D/A) substituted π-conjugated organic molecules possess extremely fast nonlinear optical (NLO) response time, purely electronic in origin. This makes them promising candidates for optoelectronic applications. The use of quantum chemical approaches for prediction of the molecular NLO properties is expected to provide guidance and accelerate subsequent experimental studies. The reliable estimation of NLO properties requires quantum chemical methods that include electron correlation and large basis sets with polarization as well as diffused functions. It was shown that MP2 and coupled cluster methods can reproduce molecular hyperpolarizabilities with high accuracy. However their application to molecules of practical interest built up of more than dozen of atoms can hardly be considered a routine task.
In the present study we have utilized four well-known hybrid DFT potentials (B3LYP, B97-2, PBE0, BMK) and MP2 method with different basis sets to estimate molecular first hyperpolarizability (β) of D/A-substituted benzenes and stilbenes, listed below. The results of DFT calculations are compared to those of MP2 method and to experimental data. Three the most significant questions are addressed in this study: 1) choice of the basis set, 2) choice of the method, and 3) the accurate way to compare calculated results to each other and to the experiment.

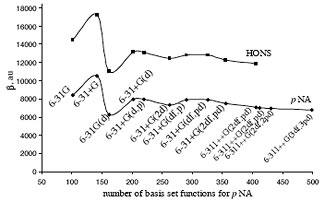
The dependence of β on the size of the basis set is plotted on Fig. 1 for pNA and HONS. While the first set of polarization and diffused functions is essential, further extension of the basis set do not change the ratio of β significantly. On these grounds we recommend 6- 31+G* basis set.
Comparison of the absolute values of hyperpolarizabilities obtained computationally and experimentally is complicated by the ambiguities in conventions and reference values used by different experimental groups. Much more tangible way is to compare the ratios of β for two (or more) given molecules of interest which were calculated at the same level of theory and measured at the same lab, using the same conventions and reference values. Coincidentally, it is the relative hyperpolarizabilities rather than absolute ones that are of the importance in the rational molecular design of efficient SHG materials. This design includes the prediction of the most promising candidates from particular homologous series which are to be synthesized and used for further investigation. In order to accomplish this goal, semiquantative level of accuracy is usually sufficient.
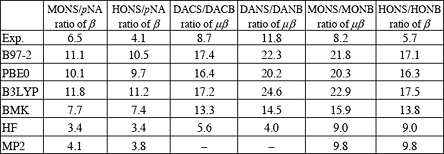
From the Table 1 one can see that MP2 values are in better agreement with experiment than DFT ones. However, even for medium-sized molecules such as HONS, MP2 and other multiconfigurational methods become impractical as a tool for systematic prediction of molecular nonlinear optical properties. At the same time DFT methods lead to systematic overestimation of β values. For BMK functional, which provides the best agreement with experiment among all DFT methods considered, overestimation is consistently close to the factor of 2. Here we propose to use the scaled BMK results for prediction of molecular hyperpolarizabilities as part of the rational design of NLO materials.